The Dichotomous Role of the Mitochondrial Permeability Transition Pore
The mitochondrion is a dynamic organelle that maintains cellular homeostasis by generating high-energy phosphates, buffering intracellular calcium, and modulating oxidative stress 1. However, mitochondria are also capable of mediating programmed cell death and necrosis 1. Central to many of these regulatory functions is the mitochondrial permeability transition pore (mPTP). Mounting evidence suggests that permeability transition (opening of the pore) within mitochondria plays a pathological role in a variety of cardiac disease processes and neurodegenerative states while physiological regulation of the mPTP has recently been shown to be important for cellular development and differentiation (Figure)2. Thus, the mPTP plays a dichotomous role in health and disease.
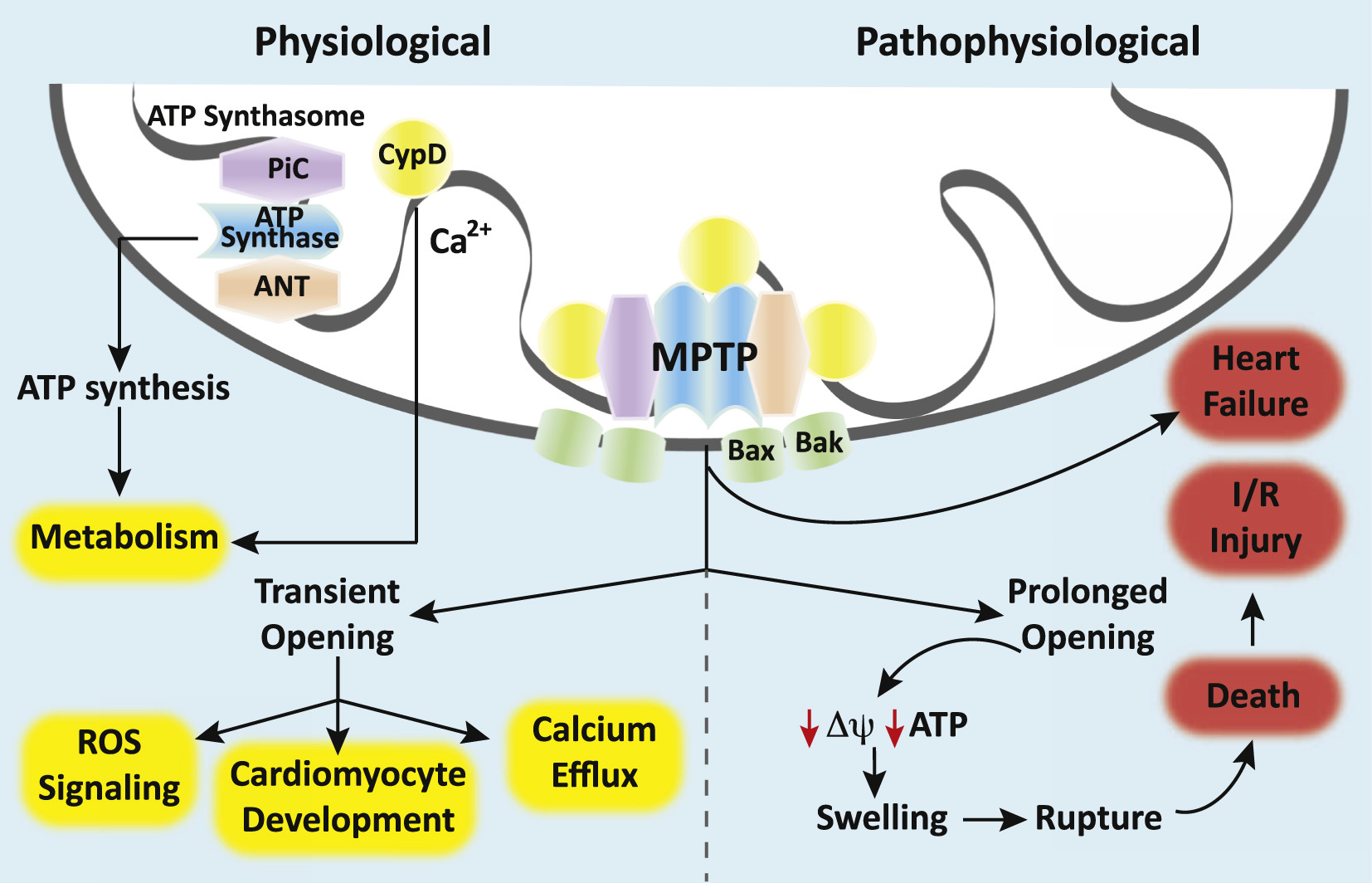
The mPTP is a voltage-gated, non-specific, non-selective mega-channel present on the inner mitochondrial membrane 2,3. Although the presence of the pore and its functional importance have been well established, its exact proteinaceous molecular identity remains elusive. Our understanding of the components that make up the mPTP has evolved over time and genetic-based studies have helped to better define the pore-forming structures and pore regulators 4. Currently, the ATP synthase (Complex V of the electron transport chain [ETC]) is believed to form the putative core component of the mPTP while the adenine nucleotide translocator (ANT), the mitochondrial phosphate carrier (PiC), and cyclophilin D (CypD) are believed to serve as pore regulators 4. Some have proposed that the c-subunit ring of the ATP synthase is the pore while others have suggested that the mPTP forms between ATP synthase dimers 3,5. CypD, a mitochondrial matrix protein with peptidyl-prolyl cis-trans-isomerase activity, promotes mPTP opening by facilitating a calcium-triggered conformational change in the pore 6. Calcium and reactive oxygen species (ROS) facilitate pore opening while adenine nucleotides, low pH, magnesium, cyclosporine A (CsA) and sanglifehrin A ([CypD] inhibitors) inhibit the mPTP 4.
High-conductance opening of the mPTP permits molecules of up to ~1.5 kD in size to pass into the mitochondrial matrix while low-conductance opening restricts permeability to molecules with a molecular mass of < 0.3 kD 2,7. The resultant inner membrane permeability can have consequences for the mitochondrial membrane potential, calcium handling, superoxide production, ATP synthesis, and cell viability 2. Uncontrolled, high-conductance opening of the mPTP causes collapse of the mitochondrial membrane potential, rapid uncoupling of oxidative phosphorylation, swelling of mitochondria, and ultimately leads to apoptosis or necrosis 2,3,7. This pathological mPTP opening is thought to underlie ischemia-reperfusion injury in the heart and other organ systems and contributes to the mitochondrial dysfunction and oxidative stress seen in neurodegenerative disorders such as Huntington’s, Alzheimer’s, and Parkinson’s disease (Figure)2. Furthermore, mPTP opening is known to play a role in the pathophysiology of traumatic brain injury (TBI) 2. Thus, the mPTP represents an important therapeutic target for a wide variety of disease processes.
In contrast to pathological opening, physiological opening of the mPTP is a regulated and controlled process important for homeostasis and cellular maturation (Figure)2. Transient, low-conductance opening of the pore has been shown to result in calcium efflux, suggesting a role in calcium regulation 2. In addition, controlled mPTP opening has been shown to result in physiological ROS signaling 2. Transient pore opening results in proton leak, a brief decline in mitochondrial membrane potential, an increase in electron flux through the ETC and a short burst or “flash” of superoxide 2. These superoxide flashes are thought to be important during cellular maturation and may mediate the metabolic switch from aerobic glycolysis to oxidative respiration in differentiating stem cells 8. Closure of the mPTP also appears to be an important process in developing cells. In embryonic cardiomyocytes, the ETC is immature and energy is known to be produced primarily via anaerobic glycolysis 2,9. During this developmental period, the cardiomyocyte mPTP is wide open, mitochondrial membrane potential is low, and ROS levels are high 2,9. Closure of the mPTP couples oxidative phosphorylation, increases the mitochondrial membrane potential, reduces oxidative stress, and induces cardiomyocyte differentiation and mitochondrial maturation 10. In the developing nervous system, neural progenitor cells also undergo a metabolic switch from glycolysis to aerobic respiration during differentiation 11. Emerging evidence suggests that the mPTP plays a role in this metabolic reprogramming to suppress neural progenitor cell proliferation and promote differentiation 8. Further evidence suggests a role for the mPTP in the differentiation of other types of cells, such as hematopoietic and vascular progenitor cells and natural killer cells 10. Therefore, developmental regulation of the mPTP is important in a variety of cells, tissues, and organ systems.
With regard to anesthetics, a number of the agents we commonly use in daily practice have been shown to impact open probability of the mPTP. Most studies have evaluated the effect of anesthetics on the mPTP in the context of ischemia-reperfusion in a variety of different cell and tissue types. Propofol, for example, has been shown to prevent reperfusion injury by inhibiting mPTP opening and calcium-induced permeability transition in rodent brain, cardiomyoctyes, and hepatocytes 12-14. Isoflurane prevents permeability transition during the reperfusion phase (postconditioning) in the rodent heart and brain, conferring cytoprotection following ischemia-reperfusion 15-17. Interestingly, anesthetic preconditioning has also been shown to promote cellular survival by preventing subsequent activation of the mPTP 18. A number of potential mechanisms of anesthetic-mediated cellular protection have been proposed by investigators and include roles for CypD, calcium, and ROS signaling 19. However, the exact mechanisms remain elusive. Importantly, detailed rigorous investigation of how various anesthetics interact with the mPTP is also lacking. An obvious barrier to our full understanding is the fact that the exact proteinaceous molecular identity of the mPTP channel is unknown. These gaps, however, represent opportunities for anesthesiologist-scientists to contribute to the literature and enhance our knowledge. Thus, future investigation should focus on attempts to identify the molecular components of the mPTP, determine if anesthetic agents bind to and modulate the pore itself, and develop novel therapeutics that target the mPTP in various disease processes. Anesthesiologists and intensivists should be aware of this channel’s biological activity and recognize that mPTP research is a fertile area for basic scientific investigation. Because the mPTP is important in health and disease, future research is likely to shed greater light on the pathophysiology of permeability transition in many different disease processes relevant to our field and elucidate the role of the pore in development and aging.
References
- Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012; 148:1145-59.
- Pérez MJ, Quintanilla RA. Development or disease: duality of the mitochondrial permeability transition pore. Dev Biol. 2017; 426:1-7.
- Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park HA, Licznerski P, Li H, Nabili P, Hockensmith K, Graham M, Porter GA Jr, Jonas EA. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc Natl Acad Sci U S A. 2014; 111:10580-5.
- Kwong JQ, Molkentin JD. Physiological and pathological roles of the mitochondrial permeability transition pore in the heart. Cell Metab. 2015; 21:206-214.
- Bernardi P. The mitochondrial permeability transition pore: a mystery solved? Front Physiol. 2013; 4:95.
- Javadov S, Karmazyn M, Escobales N. Mitochondrial permeability transition pore opening as a promising therapeutic target in cardiac diseases. J Pharmacol Exp Ther. 2009; 330:670-8.
- Javadov S, Kuznetsov A. Mitochondrial permeability transition and cell death: the role of cyclophilin d. Front Physiol. 2013; 4:76.
- Hou Y, Ouyang X, Wan R, Cheng H, Mattson MP, Cheng A. Mitochondrial superoxide production negatively regulates neural progenitor proliferation and cerebral cortical development. Stem Cells. 2012; 30:2535-47.
- Beutner G, Eliseev RA, Porter GA Jr. Initiation of electron transport chain activity in the embryonic heart coincides with the activation of mitochondrial complex 1 and the formation of supercomplexes. PLoS One. 2014; 9:e113330.
- Hom JR, Quintanilla RA, Hoffman DL, de Mesy Bentley KL, Molkentin JD, Sheu SS, Porter GA. Jr. The permeability transition pore controls cardiac mitochondrial maturation and myocyte differentiation. Dev Cell. 2011; 21:469-78.
- Zheng X, Boyer L, Jin M, Mertens J, Kim Y, Ma L, Ma L, Hamm M, Gage FH, Hunter T. Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. Elife. 2016; 5. pii: e13374.
- Morota S, Månsson R, Hansson MJ, Kasuya K, Shimazu M, Hasegawa E, Yanagi S, Omi A, Uchino H, Elmér E. Evaluation of putative inhibitors of mitochondrial permeability transition for brain disorders--specificity vs. toxicity. Exp Neurol. 2009; 218:353-62.
- Sztark F, Ichas F, Ouhabi R, Dabadie P, Mazat JP. Effects of the anaesthetic propofol on the calcium-induced permeability transition of rat heart mitochondria: direct pore inhibition and shift of the gating potential. FEBS Lett. 1995; 368:101-4.
- Zhao G, Ma H, Shen X, Xu GF, Zhu YL, Chen B, Tie R, Qu P, Lv Y, Zhang H, Yu J. Role of glycogen synthase kinase 3β in protective effect of propofol against hepatic ischemia-reperfusion injury. J Surg Res. 2013; 185:388-98.
- Ge ZD, Pravdic D, Bienengraeber M, Pratt PF Jr, Auchampach JA, Gross GJ, Kersten JR, Warltier DC. Isoflurane postconditioning protects against reperfusion injury by preventing mitochondrial permeability transition by an endothelial nitric oxide synthase-dependent mechanism. Anesthesiology. 2010; 112:73-85.
- Pravdic D, Mio Y, Sedlic F, Pratt PF, Warltier DC, Bosnjak ZJ, Bienengraeber M. Isoflurane protects cardiomyocytes and mitochondria by immediate and cytosol-independent action at reperfusion. Br J Pharmacol. 2010; 160:220-32.
- Zhao P, Ji G, Xue H, Yu W, Zhao X, Ding M, Yang Y, Zuo Z. Isoflurane postconditioning improved long-term neurological outcome possibly via inhibiting the mitochondrial permeability transition pore in neonatal rats after brain hypoxia-ischemia. Neuroscience. 2014; 280:193-203.
- Sepac A, Sedlic F, Si-Tayeb K, Lough J, Duncan SA, Bienengraeber M, Park F, Kim J, Bosnjak ZJ. Isoflurane preconditioning elicits competent endogenous mechanisms of protection from oxidative stress in cardiomyocytes derived from human embryonic stem cells. Anesthesiology. 2010; 113:906-16.
- Teixeira G, Chiari P, Fauconnier J, Abrial M, Couture-Lepetit E, Harisseh R, Pillot B, Lacampagne A, Tourneur Y, Gharib A, Ovize M. Involvement of Cyclophilin D and Calcium in Isoflurane-induced Preconditioning. Anesthesiology. 2015; 123:1374-84.
Authors


Categories
- Announcements
- Annual Meeting
- ASA News
- AUA: Then and Now
- Call for Articles
- Clinical Research Consortium
- Communications & Website Committee
- DEI
- EAB Report
- Featured Articles
- In Memoriam
- LAB Report
- Learning from Setbacks
- MEB
- Member News
- Mentoring
- Nominations
- President's Message
- SAB Report
- Scholars’ Program
- Statements
- Strategic Planning
- Submission Guidelines
- Wellness
Current Newsletter

Past Newsletters
News & Media







AUA’s EAB and LAB hosted the inaugural presentation of three new awards at AUA’s 2022 Virtual Annual Meeting. Meet the winners!
Publications of Interest
Fiftieth Anniversary of Uncovering the Tuskegee Syphilis Study: The Story and Timeless Lessons
Martin J. Tobin 1,2
1-Division of Pulmonary and Critical Care Medicine, Hines Veterans Affairs Hospital, Hines, Illinois; and -Stritch School of Medicine, Loyola University of Chicago, Maywood, Illinois
Improving Representation in Clinical Trials and Research: Building Research Equity for Women and Underrepresented Groups (2022) (National Academies of Medicine)
Kirsten Bibbins-Domingo and Alex Helman, Editors


